Komisja Europejska chce dać producentom czas na dostosowanie się do zmienionych norm bezpieczeństwa dotyczących wyrobów diagnostycznych, ponieważ wolniejsze niż oczekiwano przejście na nowe ramy stwarza ryzyko niedoborów.
Przegląd unijnych ram dotyczących wyrobów medycznych przeprowadzony w 2012 r. miał na celu zmianę niektórych przestarzałych przepisów z lat 90. XX w. oraz poprawę bezpieczeństwa i dostępności tych produktów.
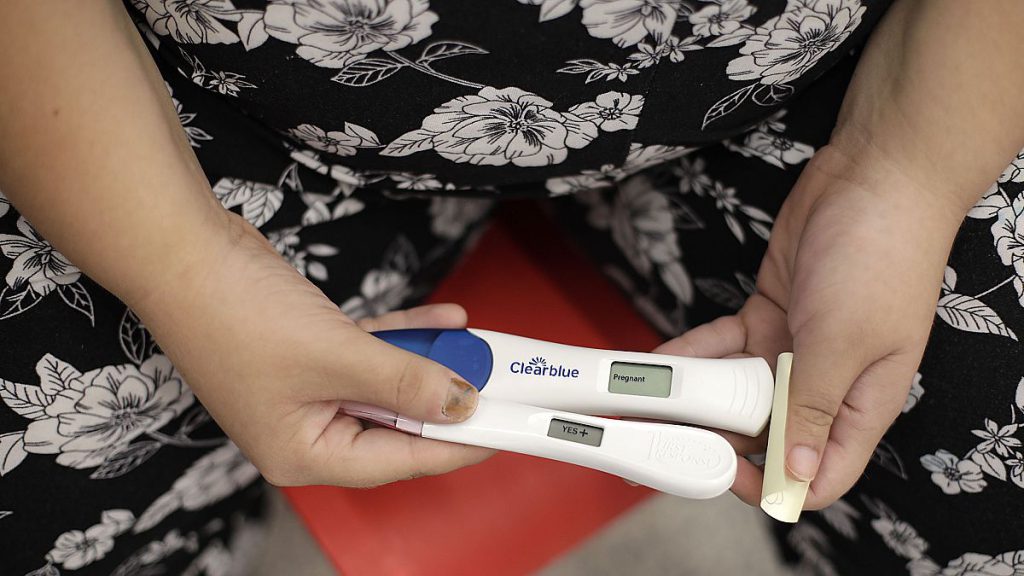
W 2017 r. zatwierdzono szczegółowe przepisy dotyczące wyrobów medycznych do diagnostyki in vitro, począwszy od glukometrów do monitorowania cukrzycy po zestawy ciążowe, a także testy do wykrywania wirusów, takich jak HIV lub koronawirus.
Nowe wymagania dotyczące diagnostyki medycznej in vitro formalnie mają obowiązywać od maja 2025 r. dla narzędzi wysokiego ryzyka i od maja 2027 r. dla narzędzi niskiego ryzyka.
Jednakże w miarę zbliżania się ostatecznych terminów wdrożenie tych zasad okazało się trudne – szacuje się, że złożono około 15 000 wniosków o zezwolenie na nowe produkty i ponowną certyfikację produktów już wprowadzonych do obrotu.
W rozporządzeniu przedstawionym we wtorek (23 stycznia) organ wykonawczy UE zdecydował się zabezpieczyć, zapewniając producentom sprzętu do diagnostyki in vitro dodatkowe dwuipółletnie przedłużenie w celu dostosowania się do nowych przepisów.
Dane zebrane przez Komisję wykazały, że operatorzy rynku nie zdołali podjąć na czas wszystkich niezbędnych kroków, aby wprowadzić do obrotu swoje urządzenia, w przypadku których obecnie istnieje ryzyko wycofania z obrotu lub utraty dostępności.
„Wniosek zapewni ulgę sektorowi, bez uszczerbku dla bezpieczeństwa i opieki nad pacjentami” – powiedziała unijna komisarz ds. zdrowia Stella Kyriakides, podkreślając, że konieczne są natychmiastowe działania, aby poprawić dostępność tych wyrobów medycznych.
Urzędnik UE powiedział TylkoGliwice, że nie jest to „czek in blanco” dla firm, ponieważ przedłużenie podlega pewnym warunkom i że producenci muszą „również wykazać chęć przejścia na nowe przepisy”.
Znalezienie przyczyn źródłowych
Nie jest to pierwsza zmiana w pierwotnych planach wdrożenia nowych ram, ponieważ w styczniu 2023 r. zaproponowano również dłuższe przejście na inne typy wyrobów medycznych.
Unijne stowarzyszenie producentów wyrobów medycznych MedTech Europe podkreśliło w oświadczeniu, że ten dodatkowy czas należy wykorzystać „na zidentyfikowanie blokad i naprawienie istniejących problemów”.
Wraz z rozporządzeniem Komisja ogłosiła, że w tym roku rozpocznie prace przygotowawcze do ukierunkowanej oceny dotychczas wyboistego prawodawstwa dotyczącego wyrobów medycznych.
„W przyszłości jesteśmy zdeterminowani przeanalizować podstawowe przyczyny spowalniające przejście i zobowiązujemy się do podjęcia odpowiednich działań” – powiedział Kyriakides.
Za jedną z tych przyczyn uważa się brak organizacji jednostek notyfikowanych, czyli tych wyznaczonych przez państwa członkowskie do oceny zgodności produktu przed wprowadzeniem wyrobów medycznych na rynek.
Obecnie istnieje jedynie 12 organów zajmujących się diagnostyką in vitro, a w przygotowaniu jest osiem kolejnych – Komisja uważa tę liczbę za wystarczającą, aby zaspokoić rosnące zapotrzebowanie.
Liczba jednostek notyfikowanych to jednak tylko jeden z problemów. „Największym problemem jest gotowość producentów, co stanowi szczególne wyzwanie dla MŚP, które są największymi producentami tych urządzeń” – powiedział urzędnik Komisji.
Proponowanemu rozporządzeniu towarzyszą pewne inicjatywy o charakterze nielegislacyjnym, których celem jest zapewnienie dodatkowego wsparcia i wytycznych ułatwiających przejście.
We wniosku uwzględniono również wymóg uprzedniego powiadomienia, zgodnie z którym producenci i operatorzy zostali poproszeni o powiadomienie właściwych organów lub osobistych użytkowników wyrobów z sześciomiesięcznym wyprzedzeniem o możliwej przerwie w dostawach.
Wszelkie wymagania nakładane na producentów dotyczące powiadamiania o zaprzestaniu produkcji wyrobów o znaczeniu krytycznym powinny być możliwie proste, aby uniknąć nakładania dodatkowych kosztów, stwierdziła MedTech Europe w oświadczeniu.